※ 研究助成に事業名を追記しました。(2026年1月9日)
発表のポイント
◆インフルエンザウイルス集団の内部に潜む遺伝子多様性を、単一RNA分子レベルで高精度に計測できる新しいゲノム解析法を確立しました。
◆ユニーク分子識別子を用いて、シーケンス由来の誤りを劇的に低減し、インフルエンザウイルス集団内の変異分布を測定することに成功しました。
◆インフルエンザのみならず、多様なRNAウイルスの進化メカニズムの理解や、AIを用いた変異予測・ワクチン株選定の高度化に貢献することが期待されます。

感染し増殖したインフルエンザウイルスゲノムにはどこにどれくらい変異が入るのか?
概要
東京大学大学院工学系研究科の玉尾研二大学院生、東京大学大学院工学系研究科(兼)プラネタリーヘルス研究機構 野地博行教授、田端和仁准教授の研究グループは、インフルエンザウイルス集団の内部に潜む遺伝子多様性を、単一RNA分子レベルで高精度に計測できる新しいゲノム解析法を確立しました。
本研究では、ウイルスRNAから合成したDNAにユニーク分子識別子(UMI)(注1)を用いて、シーケンス由来の誤りを劇的に低減しました。その結果、1塩基当たり10-5程度という極めて低い誤り率で、インフルエンザウイルス集団内の変異分布を測定することに成功しました。
この手法を、1個のインフルエンザウイルス粒子から増殖させたウイルス集団に適用したところ、同じ株由来であっても、集団中には1つの配列を中心として多数の少数変異体が広がって存在していること、その一部はランダム変異では説明できない頻度で蓄積していることが分かりました。さらに、情報理論に基づく解析によって、こうした多様性がウイルスの進化ポテンシャルを定量的に評価できることを示しました。
本成果は、インフルエンザのみならず、多様なRNAウイルスの進化メカニズムの理解や、AIを用いた変異予測・ワクチン株選定の高度化に貢献することが期待されます。
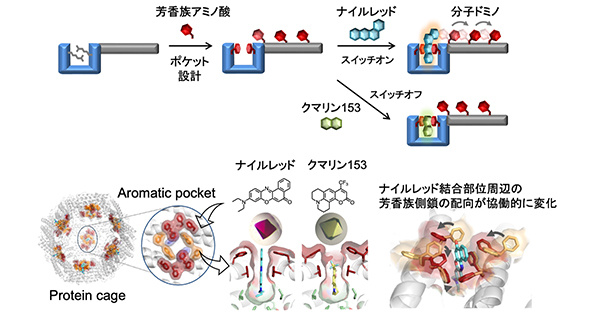
 図1:本研究の概略図
図1:本研究の概略図
発表内容
背景
インフルエンザウイルスは、エラー率の高いRNA依存性RNAポリメラーゼを用いて増殖するため、1人の感染者の体内だけでも非常に多様な変異体が共存していることが知られています。このような「似ているが少しずつ異なる」ゲノムの集合は、準種(Quasi-species)(注2)と呼ばれ、ウイルスが免疫や薬剤といった環境変化に素早く適応する源泉と考えられています。準種内のゲノム配列の多様性を定量的に測定し評価する手法の開発は、実験室内での大規模なデータ収集の実現につながると考えられます。
一方で、通常のシーケンス技術では、試料調製やシーケンサーに由来する技術的エラーが避けられず、特に0.1%以下といった低頻度変異を真に「生物学的な変異」として見分けることが困難でした。そのため、ウイルス集団内に実際にどのような変異がどの頻度で存在しているのか、またそれが将来の変異株出現とどう関わるのかを、定量的に評価することは大きな課題でした。
インフルエンザAウイルス PR8株をMDCK細胞(注3)に極低濃度で感染させ、1個のウイルス粒子から増殖したと考えられる4つの独立集団を取得し、それぞれからウイルスRNAを抽出しました。これらのウイルスRNAに逆転写(RT)反応によるUMIの付与とPCRを施し、シーケンシング結果から上記と同様に単一分子レベルのゲノム配列を復元しました。
同時に、ウイルスゲノム様配列を持つプラスミドからT7 RNAポリメラーゼで試験管内転写したRNAについても、同じ方法でシーケンスし、技術的エラーの実測値を取得しました。
結果
(1)UMI付き単一分子シーケンスによるエラー率の大幅低減
研究グループはまず、クローン化プラスミドDNAにUMIを結合し、PacBio Revioプラットフォームでシーケンスした後、同じUMIを持つリードをまとめてコンセンサス配列を作成する手法を検証しました。その結果、UMIグループサイズ(同じUMIを持つリード数)のしきい値を3以上に設定すると、エラー率は3.81×10⁵まで低下することを示しました。
 図2:UMI分子を用いて、細胞内増加でウイルスRNAに入る本来の変異(生物学的変異)と実験操作で入る変異を区別し、加えて増幅数の偏りを元の分布に復元することができる。
図2:UMI分子を用いて、細胞内増加でウイルスRNAに入る本来の変異(生物学的変異)と実験操作で入る変異を区別し、加えて増幅数の偏りを元の分布に復元することができる。
(2)技術的エラーと生物学的変異の切り分け
たとえばPB2遺伝子では、ウイルスRNAで観測された変異率1.19×10-4に対し、試験管内転写RNAでの変異率は2.22×10-6にとどまり、検出された変異の約98%がウイルス増殖過程で生じた「生物学的変異」と推定されました。また、この高い配列決定精度から本手法が10,000分の1(0.1%)レベルの頻度で存在する変異体を高い確率で検出できることを確認しました。

図3:変異率の比較。試験管内転写に比較するとウイルス増殖過程で生じた変異が顕著に高いことがわかる。
(3)準種構造と選択の痕跡の可視化-1
UMI補正後のデータから、アミノ酸配列の多様性をShannonエントロピー(注4)を用いて評価したところ、全ての遺伝子で試験管内転写RNAよりもウイルスRNAの方が高いエントロピーを示し、単一粒子由来の集団内であっても多様なアミノ酸変異が蓄積している、すなわち高い進化ポテンシャルを秘めた変異プールが形成されていることが明らかになりました。特に、HAの抗原部位近傍では平均的な変異率よりも高い値が観測され、免疫応答との関わりが示唆されました。

図4:遺伝子ごとの多様性の高さの比較。試験管内転写RNAに比較するとウイルスゲノムRNAの多様性が顕著に高いことがわかる。
(4)準種構造と選択の痕跡の可視化-2
さらに、各遺伝子について「各塩基位置で何回変異が観測されたか」の分布P(k)を作成し、平均変異回数から導かれるポアソン分布Q(k)と比較しました。もし変異導入が完全にランダムであれば、P(k)はQ(k)に近い形になりますが、実際にはウイルスRNAではポアソン分布からの有意な逸脱が観測されました。この差をJensen–Shannon Divergence(注5)で定量化したところ、試験管内転写RNAに比べてウイルスRNAの方が大きな値を示し、一部の塩基位置では変異が選択的に蓄積していることが示唆されました。

図5:遺伝子に入る変異は偏りがあるのかを分析したグラフ。ウイルスゲノムは試験管内転写RNAと比較して顕著に高い値を示しており、ポアソン分布から有意に逸脱している、つまりランダムでない形での変異の蓄積が行われていることがわかる。
(5)将来出現し得る変異株の「予備軍」の検出可能性
競合型ロジスティック成長モデル(注6)を用いたシミュレーションから、増殖後期に集団内で発生した変異であっても、野生株より5倍以上高い増殖能を持てば最終的に0.1%以上に達して検出可能となることが示されました。これは、薬剤投与や免疫圧などの環境変化が加わった際に、どのような変異体が台頭し得るかを、実験的に予測するための基盤となります。
さらに、既知の薬剤耐性変異である PA 遺伝子 I38X 変異もプール内に出現していることが確認され、ウイルス集団の適応力の広がりの大きさがうかがわれました。
今後の展開と意義
本研究で確立した単一分子シーケンス法は、インフルエンザウイルスに限らず、エラー率の高いRNAウイルス全般の準種解析に適用可能です。特に、薬剤耐性や免疫逃避、宿主域拡大に関わる変異がどの程度の頻度で自然に生じ、そのうちどれが実際に選択されるのかを、実験室内でリアルタイムに追跡できる手法として期待されます。さらに、本研究の手法で得られる高精度な変異分布データを、機械学習やAIモデルに入力することで、パンデミックを引き起こし得る変異パターンの早期予測などの社会的な貢献が期待されます。
感染症の流行は、地球環境や生態系の影響、人の都市社会の問題までもが複雑に絡み合って起こっています。
本成果は感染症の流行を理解する基礎的な成果であり、上記のようなプラネタリーヘルスに資する成果です。
〇関連情報:
プラネタリーヘルス研究機構
https://riph.adm.u-tokyo.ac.jp/
発表者・研究者等情報
東京大学 大学院工学系研究科
玉尾 研二 博士課程
東京大学 大学院工学系研究科(兼)プラネタリーヘルス研究機構
野地 博行 教授
田端 和仁 准教授
論文情報
雑誌名:eLife
題 名:Heterogeneity of Genetic Sequence within Quasi-species of Influenza Virus Revealed by Single-Molecule Sequencing
著者名:Kenji Tamao, Hiroyuki Noji, Kazuhito V Tabata
DOI:https://doi.org/10.7554/eLife.108882.1
URL:https://elifesciences.org/reviewed-preprints/108882
研究助成
本研究は科学技術振興機構(JST)戦略的創造研究推進事業 CREST「AI が先導するオートメーションタンパク質工学の創出」(No. JPMJCR22N2)、日本医療研究開発機構(AMED)SCARDA「ワクチン開発のための世界トップレベル研究開発拠点の形成事業」ワクチン開発のための世界トップレベル研究開発拠点群 東京フラッグシップキャンパス(東京大学新世代感染症センター)2024-2025 年度UTOPIA AI 研究発掘プログラム(No. JP223fa627001)の支援を受けて行われました。
用語解説
(注1)ユニーク分子識別子(UMI:Unique Molecular Identifier):数塩基〜十数塩基程度の短いDNA配列からなる分子バーコード。ライブラリ調製の増幅前の段階で各RNA/DNA分子に一対一対応で付加することで、PCRで生じたコピーを区別し、元の分子数の正確なカウントやシーケンスエラー補正を可能にする。
(注2)準種(Quasi-species):高い変異率で増殖するRNAウイルスなどで見られる、ある配列を中心に少しずつ配列の異なる多数のゲノム変異体が集団として共存している状態。免疫回避や薬剤耐性獲得など高速な進化の源になると考えられている。
(注3)MDCK細胞:Madin-Darby Canine Kidneyの略で、イヌ腎臓由来の上皮様培養細胞株。インフルエンザウイルスをはじめとする多くのヒトインフルエンザウイルスの分離・増殖に広く用いられており、ワクチン製造用の細胞基材としても利用されている。
(注4)Shannonエントロピー:情報理論で導入された量で、ある確率分布の「不確実さ」や「多様性」の大きさを数値化したもの。取り得る状態の種類が多く、かつそれぞれが均等な確率で現れるほどエントロピーは高くなる。
(注5)Jensen–Shannon Divergence(JSD):2つの確率分布がどれだけ異なっているかを測る統計量。0に近いほど分布が似ており、1に近いほど違いが大きい。非対称で値が発散し得るKLダイバージェンスと比べ、JSDは常に有限で対称な「距離」として扱える。
(注6)ロジスティック成長モデル:初期には指数関数的に増えるが、資源が限られると成長が頭打ちになる個体群の増殖を記述する数理モデル。初期条件として初期個体数、増殖率、環境収容力を与えることで、個体数の時間変化をシミュレーションすることができる。競合型モデルでは野生型と変異型で共通の資源を奪い合う形でシミュレーションを行う。
プレスリリース本文(2026年1月9日訂正版):PDFファイル
光合成反応における光損傷と修復のメカニズム解明 ~傷ついたタンパク質を見つけて分解するしくみを明らかに~
神経抑制に役立つタンパク質の巧みな光応答メカニズムを解明