Authors
Ena Tomoda, Asuteka Nagao, Yuki Shirai, Kana Asano, Takeo Suzuki, Brendan J Battersby, Tsutomu Suzuki
Abstract
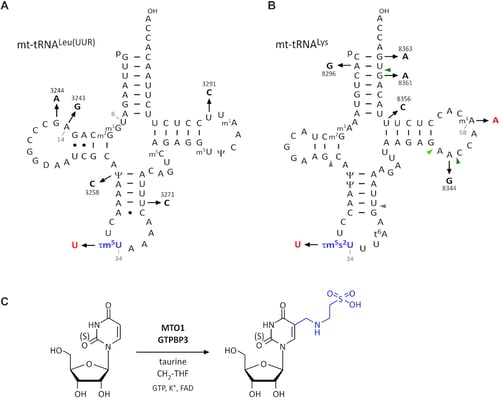
Mutations in mitochondrial (mt-)tRNAs frequently cause mitochondrial dysfunction. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), and myoclonus epilepsy associated with ragged red fibers (MERRF) are major clinical subgroups of mitochondrial diseases caused by pathogenic point mutations in tRNA genes encoded in mtDNA. We previously reported a severe reduction in the frequency of 5-taurinomethyluridine (τm5U) and its 2-thiouridine derivative (τm5s2U) in the anticodons of mutant mt-tRNAs isolated from the cells of patients with MELAS and MERRF, respectively. The hypomodified tRNAs fail to decode cognate codons efficiently, resulting in defective translation of respiratory chain proteins in mitochondria. To restore the mitochondrial activity of MELAS patient cells, we overexpressed MTO1, a τm5U-modifying enzyme, in patient-derived myoblasts. We used a newly developed primer extension method and showed that MTO1 overexpression almost completely restored the τm5U modification of the MELAS mutant mt-tRNALeu(UUR). An increase in mitochondrial protein synthesis and oxygen consumption rate suggested that the mitochondrial function of MELAS patient cells can be activated by restoring the τm5U of the mutant tRNA. In addition, we confirmed that MTO1 expression restored the τm5s2U of the mutant mt-tRNALys in MERRF patient cells. These findings pave the way for epitranscriptomic therapies for mitochondrial diseases.
Nucleic Acids Research: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkad139/7076465

Molecular mechanism on forcible ejection of ATPase inhibitory factor 1 from mitochondrial ATP synthase
Complete chemical structures of human mitochondrial tRNAs
