Share this
High-Throughput Investigations of Topological and Nodal Superconductors
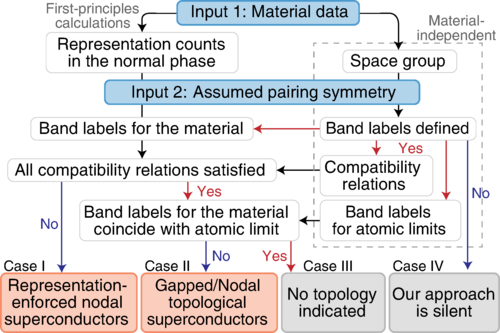
High-Throughput Investigations of Topological and Nodal Superconductors
2022/07/07
RadonPy: automated physical property calculation using all-atom classical molecular dynamics simulations for polymer informatics
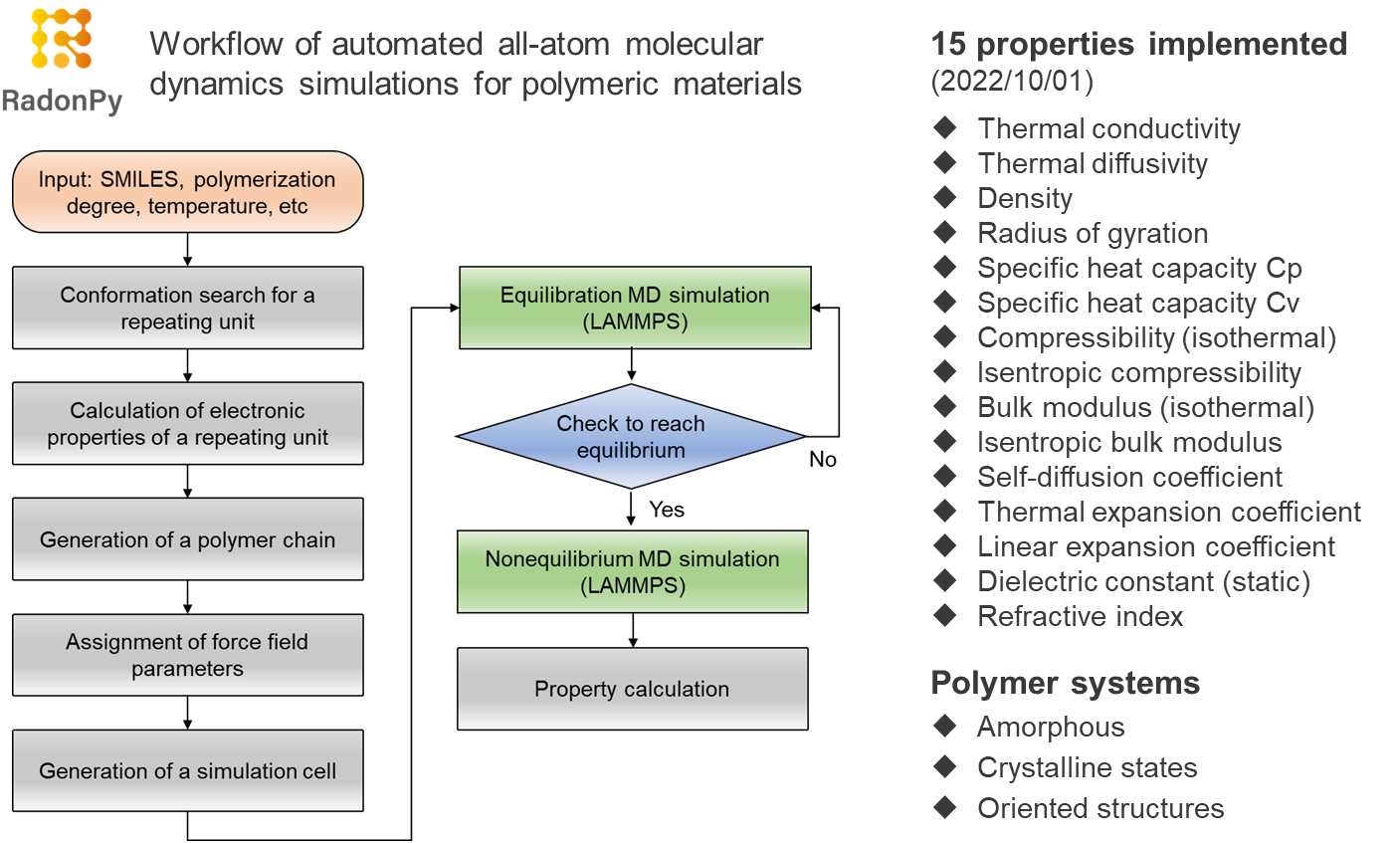
RadonPy: automated physical property calculation using all-atom classical molecular dynamics simulations for polymer informatics
2022/11/09
ARIM-mdx Data System: A New Data Platform Revolutionizing Materials Research with 900+ Active Users in Japan
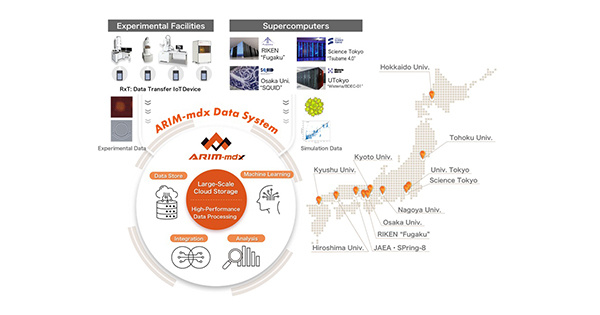
ARIM-mdx Data System: A New Data Platform Revolutionizing Materials Research with 900+ Active Users in Japan
2024/12/13